Scientists and programmers convert science to algorithms, which are then implemented as programs. The science and algorithms are of interest to scientists, but the implementation of algorithms as programs is much less interesting to scientists, who want to obtain correct, efficient, flexible implementations as quickly as possible.
The POOMA (Parallel Object-Oriented Methods and Applications) Toolkit, developed at Los Alamos National Laboratory, provides high-level C++ tools to rapidly implement many scientific algorithms as C++ programs. The toolkit makes scientific tools (e.g., arrays, fields, and data-parallel expressions) available while hiding the underlying computer science.
In this article, I will describe the POOMA Toolkit and show how POOMA eases the task of translating scientific algorithms into programs. Then, I will illustrate how to use POOMA by implementing a two-dimensional diffusion algorithm. A few changes convert the program to run on a computer with thousands of processors. I conclude by describing how the open-source POOMA Toolkit can make scientific programming easier and more efficient.
Easy Translation from Algorithms to Programs
POOMA’s concepts and syntax mimic notation used to describe scientific algorithms, so scientific programs written using POOMA are:
- Easier to write.
- Significantly shorter.
- Easier to debug.
- More likely to be correct.
For example, a mathematical formula x = 3sin((v–w)/p) – 4.2cos(w2) can be directly converted to C++ code:
x = 3.0 * sin((v - w) / M_PI)
- 4.2 cos(w * w)
where v, w, and x are scalar values or vectors or matrices or tensors or even containers of values. Error-prone memory allocation and deallocation is eliminated because POOMA containers automatically allocate and deallocate memory to store data. Furthermore, containers are first-class objects, so they can be used as function arguments and copied as simply as if they were built-in scalar values.
Because the toolkit automatically supplies all interprocessor communication, programmers familiar with writing sequential programs can easily write POOMA programs that run on thousands of processors. Since the same executable can run on any number of processors, many POOMA programs are developed and debugged on uniprocessor workstations and then run on multiprocessor computers without any need for additional debugging.
All these benefits together speed the process of converting algorithms into programs so scientists can spend their time on what is important: the science. For example, a team of four physicists at Los Alamos National Laboratory recently used POOMA to implement an extensive set of physics simulations, averaging just two days to translate each scientific paper into a program.
POOMA Concepts
Three concepts you’ll need to understand in order to work with POOMA are:
- Containers: POOMA containers support data access and storage. POOMA Arrays extend the functionality of C++ arrays by supporting multiple dimensions and data-parallel expressions. Other POOMA containers include Fields (which support field-based scientific computations where values are located at positions within grid cells), Vectors, Matrixes, and Tensors.
- Computation modes: computation modes describe the syntax used to perform computations on data stored in containers. C and C++ programmers are familiar with element-wise computation mode, where each container value (a.k.a. element) is individually accessed. Many familiar mathematical operations require an alternative data-parallel mode, where multiple values are simultaneously accessed (see sidebar, “POOMA Data-Parallel Syntax”). Other computation modes include stencils (illustrated in the program below) and relational computations. Relational computation implements lazy evaluation, where one container’s values depend on several other containers’ values.
- Computation environment: the computation environment indicates the machine and operating system that will run a program. Sequential computation using just one thread of execution is familiar to most programmers. POOMA also supports distributed programming using the SPMD (single-program, multiple-data) model, which is particularly appropriate for scientific programs. Any number of processors execute the same program, each using different data.
The POOMA Toolkit
The POOMA Toolkit is a general-purpose library designed to implement scientific and technical programs. It has been used to simulate advection equations used in modeling weather, simulate diffusion processes such as the neutron diffusion program (available for download at <www.cuj.com/code>) described in this article, and solve systems of linear equations. It could be used to model oil reservoirs or predict the values of financial options. It has even been used to implement Conway’s Game of Life. These programs have been run on single-processor desktop computers and on massively parallel computers with thousands of processors.
The POOMA Toolkit provides a wide variety of tools. To store groups of values, it provides single- and multi-dimensional array and field containers. As anyone who writes scientific or technical programs knows, arrays (or grids) of values form the heart of these programs. The Field container extends this concept to three-dimensional space, so values are not only stored but the distances between them can be computed. Values within containers can be individually manipulated, but data-parallel operations operate collectively on the values in ways analogous to Fortran 90. Stencils support local computations such as in Conway’s Game of Life where the life or death of a cell depends on its neighboring values. The toolkit provides vector, matrix, and tensor classes and mathematical operations. It has been built to support multiprocessor computation and communication with a minimum of fuss. Programmers need only specify how each container’s data should be distributed among the processors, and POOMA automatically ensures all necessary interprocessor communication occurs. Thus, the same oil reservoir simulation program might simulate a small field using a desktop computer and simulate an elephant field using a cluster of computers.
Neutron Diffusion Algorithm
POOMA eases the translation of scientific algorithms into programs, and I illustrate this with a two-dimensional diffusion simulation. Diffusion problems occur frequently in science, and I present one based on 1943 lectures at the newly created Los Alamos National Laboratory, which developed the POOMA Toolkit fifty years later.
To introduce the problem of making an atomic bomb to the incoming scientists and engineers, Robert Serber described the neutron diffusion process that powers an atomic bomb [1]. The rate of increase of free neutron density N in a supercritical nuclear mass is modeled by the diffusion equation:
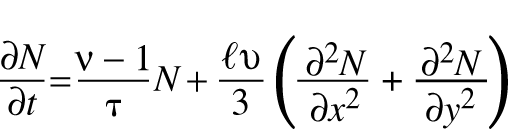
where n, t,  , and u are constants.
, and u are constants.
To simulate the diffusion, time and two-dimensional space are discretized, yielding:
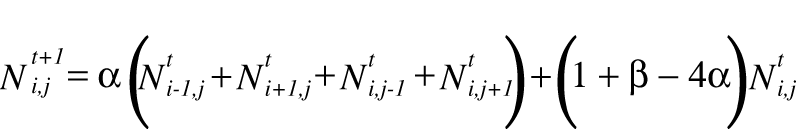
where ![]() indicates the number
of neutrons at time t at grid position (i,j), a is a diffusion
constant in the range (0,0.5), and b indicates the number of neutrons
created by each fission. In words, the equation indicates that the number of
the neutrons at grid position (i,j) at time step t + 1 is dependent
on the number of neutrons at time step t at grid position (i,j)
and its neighboring cells.
indicates the number
of neutrons at time t at grid position (i,j), a is a diffusion
constant in the range (0,0.5), and b indicates the number of neutrons
created by each fission. In words, the equation indicates that the number of
the neutrons at grid position (i,j) at time step t + 1 is dependent
on the number of neutrons at time step t at grid position (i,j)
and its neighboring cells.
The recurrence equation, as presented, could easily be implemented, but its local nature facilitates use of a stencil, which is used by many scientific algorithms. A stencil is a rectangular tile that, when applied to a grid, yields a value. (For more information on stencils, see the sidebar, “Stencils.”) Figures 1 and 2 illustrate how a stencil is applied to a grid. A stencil contains coefficients for a particular cell (indicated by solid lines in Figure 1) and neighboring cells. When applying the stencil to a grid position, the stencil’s coefficients are multiplied by corresponding grid values and then combined (e.g., added). Figure 2 indicates the same stencil can be applied to various grid positions even if the stencil applications overlap.
Rewriting the recurrence equation in terms of the stencil in Figure 1 yields:
![]()
meaning the new neutron density N equals the application of the stencil at grid position (i,j).
Free neutrons at the edge of the mass escape without causing fission, so the neutron density on the edges of our mass is zero, yielding a boundary condition. To ease notation, the program will simulate a square mass, rather than a circular mass. Initially the simulation will have no free neutrons present except at the center of the mass.
Algorithm 1 summarizes the simulation. After establishing initial conditions, a series of time steps are simulated. During each time step, all free neutron densities are updated using the stencil, boundary conditions are again applied, and the current simulation values are inspected if desired.
Uniprocessor Program
Now that the algorithm is decided, I will translate it to a C++ program using the POOMA Toolkit (i.e., a “POOMA program”). POOMA supports stencils so the translation will be easy. The free neutron density will be represented by a two-dimensional array N holding floating-point numbers. To declare a n x n grid use:
Array<2> N(n,n);
The declaration creates a two-dimensional array object, as the template parameter 2 indicates. POOMA Arrays are first-class objects, automatically handling necessary memory allocation and deallocation, negating any need for explicit destruction.
Two statements implement the two initial conditions:
N = 0.0; N(n/2,n/2) = 1.0e+7;
The first, data-parallel statement assigns 0.0 to all values in N, which knows its domain size so it can iterate through all its values. The second statement assigns a nonzero neutron density to the central grid position. Note parentheses, not brackets, surround Array indices.
To create the stencil, a POOMA Stencil is instantiated with a function object. Listing 1 shows the Fisher function object class. (“Fish” is a historical word for “fission.”) A POOMA Stencil function object must have three member functions: two indicating the stencil’s size and one computing a value. As illustrated in Figure 1, the stencil extends one cell to the left of the stencil’s central position and one cell below. Thus, the function object’s lowerExtent, taking a dimension argument, returns the number of cells to the left (or below) the central position. upperExtent indicates the number of cells to the right (or above) the central position.
The stencil’s operator() computes the value from applying it to a particular grid position. Given three parameters that indicate an Array or other container C and a two-dimensional position (i,j), it reads container values and computes a value. Since the stencil in Figure 1 has five nonzero values, the computation reads five values. For example, C.read(i,j) reads the value corresponding to the stencil’s central point. Using the container’s read member function rather than syntax like C(i,j) tells the compiler the value will be read and not changed, perhaps permitting better optimization. Each value is weighted by the appropriate value and then added to yield the function’s return value.
The Fisher function object depends on its a and b parameters, which are supplied as constructor arguments. To create the stencil stencil, a Fisher object storing a and b is supplied:
Fisher fisher(alpha, beta); Stencil<Fisher> stencil(fisher);
Applying the stencil to all grid positions is simple:
N = stencil(N);
stencil is applied to every position in the two-dimensional array N. Applying a stencil to a cell on the edge of the grid causes the stencil to try to read non-existent container values, so the stencil should only be applied to interior cells. interiorDomain, declared as:
Interval<2> interiorDomain (Interval<1>(1,n-2), Interval<1>(1,n-2));
indicates these cells. The statement:
N(interiorDomain) = stencil(N, interiorDomain);
applies the stencil to only the interior cells of N, assigning the computed values to those same interior cells.
Unfortunately, this data-parallel POOMA statement is not correct. The statement is implemented one grid position at a time without storing any intermediate values. Thus, the statement has a read-write race condition. For example, both the values of N(3,3) and N(3,2) at time step t + 1 depend on N(3,3) and N(3,2) at time step t. Neither new value can be computed and assigned before the other. Doubling the number of arrays avoids the problem:
Na(interiorDomain) = stencil(Nb, interiorDomain); Nb(interiorDomain) = stencil(Na, interiorDomain);
Since each array is either read or written in each statement, the read-write race condition is avoided. The declaration of N is correspondingly changed to declare two Arrays Na and Nb.
Since only interior cells of Na and Nb are assigned values, the boundary cells’ values remain unchanged at 0.0, so the boundary condition is maintained, and no explicit code needs to be written.
The stencil application is contained with a loop counting the desired number of iterations. During any loop iteration, the neutron densities can be inspected. For example, use:
std::cout << N << std::endl;
to print all the values in N. When the program finishes, it prints the central neutron density.
To support using the POOMA Toolkit, Pooma::initialize and Pooma::finalize must be called before and after, respectively, using any POOMA code.
Listing 2 summarizes the sequential program. I encourage you to compare it with Algorithm 1. To run the program neutron-sequential, use a command like neutron-sequential 10 5 0.2. This execution has 10 grid points along each of its two dimensions, performs five time steps, and uses an a value of 0.2. a is constrained to the range (0.0, 0.5).
Multiprocessor Program
A key advantage of the POOMA Toolkit is that the same executable can run on one or thousands of processors. Thus, a scientist can use a uniprocessor workstation to design, program, and debug a program and then port to a multiprocessor with no code changes or need for more debugging.
To convert a program designed for sequential execution to one designed to run on multiple processors, one must:
- Modify the declaration of containers.
- Modify program input and output.
- Add guard statements before accesses to individual container values.
Almost all computations and program control flow remain unchanged. There is no need to add explicit communication commands. Thus, almost all of the program remains unchanged.
SPMD Model
POOMA implements a SPMD programming model. Thus, each processor runs the same program, but containers’ data can be distributed across the processors. Each processor runs the same statements but is restricted to assignments for data it owns. If an assignment requires data from other processors, the toolkit automatically issues the required communication commands to obtain the data from these processors. No programmer-specified communication syntax is required, but the program must specify how each container’s data is distributed across processors.
Figure 3 illustrates how a 6 x 6 grid might be partitioned among four processors. Grid cells, outlined with solid lines, are split into four equally sized patches, each 3 x 3. Each processor stores its patch of data and performs the computations to assign values to this data.
Computing a grid value using the stencil requires knowing its neighboring values. For grid values on an “interior” edge of a patch, this requires using the values from neighboring patches. These values could be communicated from adjacent patches, but each value might be read many times before it is changed. Thus, the values can be cached on each processor requiring their use. A guard cell is a read-only cell duplicating a value from an adjacent patch. The dashed cells in Figure 3 indicate these cells. Arrows illustrate the source locations for a few guard cells. The POOMA Toolkit automatically ensures the guard cells’ values equal their source grid values. The boundary conditions require the outermost cells have zero values, and they are not computed, so no “external” guard cells are needed.
(The careful reader may have noticed that each guard cell value is used only once before its source value is changed, so the use of guard cells in this program does not necessarily lead to a more efficient program. If, for example, the stencil computation involved all the stencils’ neighboring values rather than just those in the shape of a cross, using guard cells would reduce the necessary communication. For this program, using guard cells does not hurt the program’s efficiency since the values are used once.)
Container Declarations
For a multiprocessor program, the declarations of Arrays Na and Nb need to be modified to indicate how the data should be partitioned and how it should be mapped to processors. A partition describes how a domain should be split into pieces. The UniformGridPartition splits a domain into pieces of the same size:
UniformGridPartition<2>
partition(Loc<2>(nuProcessors,
nuProcessors),
GuardLayers<2>(1),
GuardLayers<2>(0));
The first argument indicates that applying partition to a two-dimensional domain will split each dimension into nuProcessors equally sized pieces. The second and third arguments indicate the desired number of internal and external guard cells, respectively. The template argument 2 indicates a two-dimensional partition. A UniformGridPartition partition requires that each dimension of the domain be evenly divisible into the desired number of pieces. For this reason the number n of grid points is rounded up to the nearest multiple of nuProcessors:
n = ((n+nuProcessors-1) / nuProcessors)
* nuProcessors;
Applying a partition to a domain yields a set of patches, which must be mapped to processors. For the diffusion program, the patches should be distributed among the available processors. The layout specifies this one-to-one mapping:
UniformGridLayout<2>
layout(vertDomain, partition,
DistributedTag());
The domain vertDomain to partition is declared as the cross product of two intervals:
Interval<2>(Interval<1>(0, n-1),
Interval<1>(0, n-1))
The second and third arguments specify the partition and the one-to-one mapping.
Finally, the Arrays are declared using layout as the constructor argument:
Array<2, double,
MultiPatch<UniformTag, Remote<Brick> > >
Na(layout);
Array<2, double,
MultiPatch<UniformTag, Remote<Brick> > >
Nb(layout);
The sequential declaration for Na has only one template argument because Array’s two other default arguments of double and Brick suffice. double indicates the type of values stored in Array, and Brick indicated how the data will be stored (i.e., each value should be explicitly stored). (The ability to specify data storage independent of its use, which is what the container type indicates, permits optimizations based on programmer knowledge. For example, if all the stored data frequently has the same value, a CompressibleBrick rather than a Brick can be used.)
Na’s MultiPatch<UniformTag, Remote<Brick> > declaration has four pieces:
- MultiPatch: the container’s data is distributed among multiple patches.
- UniformTag: the patches all have the same size.
- Remote<Brick>: a patch may reside on any processor in the computer.
- Brick: each patch, on a particular processor, should explicitly store all its data.
Just like High Performance Fortran [2], POOMA supports a wide variety of data distributions, but the programmer must explicitly declare how each container’s data is to be distributed. The optimal distribution depends on how the containers are used. It would be nice if POOMA automatically determined optimal distributions, but doing so remains an unsolved computer science problem.
Input and Output
In the SPMD model, many processors run the same executable on different data, so each executable instance might have its own standard input and standard output. Thus, one use of std::cout in a SPMD program may produce many statements on a terminal, up to one per processor. To avoid this flood of data, POOMA provides the Inform output stream class, which works like a std::ostream class in a multiprocessor environment. Writing to an Inform object, for example:
output << Na(n/2,n/2) << std::endl;
produces only one output regardless of the number of processors.
Declaring such an object is simple:
Inform output;
Thus, all calls to std::ostream in the sequential program should be changed to use an Inform stream. Inform streams also work for sequential programs, so POOMA programmers frequently use them rather than std::cout and std::cerr.
To avoid difficulties with program input when each processor might have its own std::cin, POOMA programs frequently use command-line arguments to pass input data. Since the sequential program already uses command-line arguments, it does not need to be modified for its multiprocessor version except that the number of processors must also be specified.
Avoiding Race Conditions
The POOMA Toolkit automatically prevents race conditions in multiprocessor programs except for one case: accessing an individual container value immediately after performing a data-parallel statement. Almost all computation in POOMA programs can be expressed using data-parallel syntax, so accesses to individual Array values are rare. When a data-parallel statement precedes accessing an individual value, a Pooma::blockAndEvaluate() statement should occur before accessing the individual value. For example, the initial conditions are specified as:
Na = Nb = 0.0; Pooma::blockAndEvaluate(); Nb(n/2,n/2) = 1.0e+7;
blockAndEvaluate ensures all processors have completed their assignments of zero before the central grid value is assigned. This prevents a race condition where the 0.0 assignments are intermixed with the assignment to the central grid position.
The blockAndEvaluate statement is only necessary for multiprocessor execution. But since POOMA programs are so easily and frequently ported to multiprocessor computers, it is good POOMA programming practice to include these statements even when writing sequential programs, where these statements have no effect.
Running the Program
The command to run a multiprocessor POOMA program depends on the computer’s operating system and messaging library. If using a MPI Library [3], the command:
mpiexec -n 9 neutron -mpi 3 18 5 0.2
runs neutron on nine processors. mpiexec -n 9 cmd is an MPI command to run cmd on nine processors. (mpiexec is not available with all MPI implementations. Check your MPI manual for the appropriate command.) neutron -mpi 3 18 5 0.2 is similar to running the sequential program except POOMA uses the -mpi option to determine the type of the communication library, and 3 is the number of processors along one dimension. Pooma::initialize strips the -mpi option from the command-line arguments before the diffusion program parses them.
MPI, the POOMA Toolkit, and the computer automatically determine which nine processors to use, how to communicate among these processors, and how to run the programs on these processors. This information need not be encoded in a POOMA program in any way.
Summary
Converting the uniprocessor neutron diffusion program to the multiprocessor version in Listing 3 requires specifying how each container’s data is distributed among all the processors, ensuring the program uses command-line arguments for input and Inform objects for output, and using blockAndEvaluate to avoid race conditions before accessing individual container values. Since POOMA programmers naturally do the latter two even when writing sequential programs, the difference between a sequential and multiprocessor program is usually only a few container declarations. The vast majority of a program, including all its computation (except blockAndEvaluate, which I assume has already been added) is the same. The sequential thought processes and control flow logic in which most programmers are trained can be used to produce POOMA programs that run on one or thousands of processors.
Efficiency
The POOMA Toolkit provides high-level scientific syntax without sacrificing efficiency. Efficiency is measured in a variety of ways:
- Programmer time: the time to write and debug the initial version of a program.
- Maintenance time: the time to maintain a program, fixing errors and adding features.
- Run time: the time to run a program.
- Software cost: the cost of software to support the program (e.g., compilers and libraries).
POOMA programs are usually up to an order of magnitude shorter than comparable C or Fortran programs because they use POOMA’s high-level scientific syntax, and there is no explicit interprocessor communication syntax. Since the time to write and debug a program usually depends superlinearly on its length, the time savings can be significant. Furthermore, scientific programmers using POOMA need not take the time to learn computer-science skills unimportant to their science (e.g., interprocessor communication protocols, how to avoid — or detect — race conditions, and how to implement lazy evaluation).
POOMA extensively uses template technology to produce code comparable to hand-written C code. For example, each data-parallel statement is translated into one loop regardless of the number of operands. The POOMA statement involving sin and cos listed above is translated into code equivalent to:
for (unsigned index = 0;
index < x.size();
++index)
x[index] = 3.0 *
sin((v[index] - w[index]) / M_PI)
- 4.2 * cos(w[index] * w[index]);
This expression-template technology used in POOMA is so powerful it is also separately available as the PETE Library [4]. Using POOMA does rely heavily on good compiler optimization, but many current C++ compilers (e.g., g++ [5] and the defunct KCC [6]) provide it.
POOMA programs can be slightly slower than hand-coded C or Fortran programs, but the ease of writing, debugging, and maintaining these significantly shorter programs usually outweighs the slight increase in running time. Profiling can reveal the most time-critical portions of a POOMA program, which can be rewritten to run more quickly.
Conclusions
POOMA uses sophisticated C++ to ease writing scientific programs. Containers appropriate for scientific programs, a wide variety of computation syntax, and easy support for writing multiprocessor programs ease translating scientific algorithms into programs, as illustrated with the neutron diffusion algorithm. These programs are easier to write and debug, require significantly fewer lines, and are easier to maintain than comparable hand-coded programs.
The POOMA Toolkit is open-source software that can be freely used, is available without payment, and can be extended to support additional specific syntax or domains.
The POOMA Toolkit and portions of a user manual are available at the CodeSourcery website (<www.codesourcery.com/pooma/pooma/>). For those wanting to know more about POOMA, Addison-Wesley will soon publish my book Scientific Computing Using Sophisticated C++, describing how using POOMA eases writing scientific programs.
Bibliography
[1] Robert Serber. The Los Alamos Primer (University of California Press, 1992).
[2] Charles H. Koelbel, David B. Loveman, and Robert S. Schreiber. The High Performance Fortran Handbook (MIT Press, 1996).
[3] The Message Passing Interface (MPI) Standard,<www-unix.mcs.anl.gov/mpi/>.
[4] Scott Haney, James Crotinger, Steve Karmesin, and Stephen Smith. “The Portable Expression Template Engine,” Dr. Dobbs Journal, October 1999, <www.acl.lanl.gov/pete>.
[5] GNU Compiler Collection, <http://gcc.gnu.org/>.
[6] KAI C++ Compiler, <http://developer.intel.com/software/products/kcc/>.
Jeffrey D. Oldham worked with the POOMA developers on POOMA v2.4. He received a Ph.D. in computer science from Stanford University and has taught at the university level. He currently works for CodeSourcery, LLC, developing software tools for scientific computing and working on compilers.